Show setup information.
knitr::opts_chunk$set(echo = TRUE)
set.seed(019911)
pacman::p_load(DiagrammeR, DiagrammeRsvg, rsvg,
install = FALSE, update = FALSE)
options(scipen=999)
knitr::opts_current$get(c(
"cache",
"cache.path",
"cache.rebuild",
"dependson",
"autodep"
))Data Availability
All files generated in this workflow can be downloaded from figshare.
File names and descriptions:
- 00_RAW/: Raw fastq files before trimming.
- 00_LOGS/: Individual log files for each step of the snakemake workflow.
- 00_TRIMMED/: Eight trimmed, compressed fastq files for each sample.
- 01_QC/: Merged forward (R1) & reverse (R2) QC’ed fastq files. QC STATS file for each sample.
qc-report.txtfile, a summary table of all QC results. - 02_FASTA/: A contig fasta file and reformat report for each assembly.
- 03_CONTIGS/: An annotated contig database for each assembly.
- 04_MAPPING/: Short read BAM files.
- 05_ANVIO_PROFILE/: Individual profile database for each sample.
- 06_MERGED/: Single merged profile database.
The contigs.db contains all taxonomic and functional annotations. The profile.db contains information on coverage, detection, etc. as well as COLLECTION information for automatic binning and manual refinement (see step 5).
The Workflow
In this section of the workflow we begin with raw, paired-end Illumina data. We will use a Snakemake (Köster and Rahmann 2012) workflow in the anvi’o (Eren et al. 2015) environment for most of the steps, though we will at time call on additional tools for specific steps. The overall structure of our workflow was modelled after the one described by Delmont & Eren on Recovering Microbial Genomes from TARA Oceans Metagenomes (Delmont et al. 2018).
Specifically, we will:
- Perform adapter trimming.
- Use the anvi’o Snakemake workflow to:
- quality filter trimmed, raw reads;
- co-assemble metagenomic sets;
- recruit reads;
- profile the mapping results;
- merge profile dbs;
- classify reads and genes;
- annotate genes;
- run HMM profiles.
- Use external tools for annotation and classification.
The main steps of this workflow are:
| Task | Tool/Description | Input |
|---|---|---|
| TRIMMING | TRIMMOMATIC to remove adaptor sequences. | raw reads |
| QUALITY-FILTERING | IU filter quality Minoche to QC trimmed reads. | trimmed reads |
| CO-ASSEMBLY | MEGAHIT to co-assemble metagenomic samples. | QC reads |
| GENE CALLING | PRODIGAL for gene calling. Contig db for results. | assembled contigs |
| TAXONOMIC ANNOTATION | KrakenUniq for short read classification. | QC reads |
| RECRUITMENT | BOWTIE2/SAMtools for mapping short reads to assembly. | QC reads, contigs db |
| PROFILING | Profile mapping results. Store in profile dbs. | BAM file |
| TAXONOMIC ANNOTATION | KAIJU & CENTRIFUGE for classification of genes. | genes (dna format) |
| FUNCTIONAL ANNOTATION | Gene annotations against Pfams, COGS, KEGG, etc. | genes (aa format) |
| MERGING | Create single merged profile db. | individual profile dbs |
Samples
We have a total of four samples from two sites and two time periods—so one sample per site per time.
- Cayo Roldan: the site that went hypoxic. WROL 1914 collected during the event and WROL 1915 collected one month after the event.
- Cayo Coral: the site that was always normoxic. WCCR 1913 collected during the event and WCCR 1916 collected one month after the event.
Trimming
Before we run the workflow we must trim Illumina adaptors. First we need to make some directories to put the output files.
mkdir 00_TRIMMED
mkdir 00_TRIMMED_UNPAIREDNext we run TRIMMOMATIC (Bolger, Lohse, and Usadel 2014) assuming the raw data is in a directory called RAW. Here is an example for a single sample (WCCR_1913). Since we ran these samples on a NextSeq, there are 4 files per paired-end. So, we have 8 files total. We need to run the command 4 times, one for each pair. Here is the command for the first pair.
trimmomatic PE -threads $NSLOTS RAW/WCCR_1913_S74_L001_R1_001.fastq.gz /
RAW/WCCR_1913_S74_L001_R2_001.fastq.gz /
00_TRIMMED/WCCR_1913_S74_L001_R1_001_trim.fastq.gz /
00_TRIMMED_UNPAIRED/WCCR_1913_S74_L001_R1_001_trim_unpaired.fastq.gz /
00_TRIMMED/WCCR_1913_S74_L001_R2_001_trim.fastq.gz /
00_TRIMMED_UNPAIRED/WCCR_1913_S74_L001_R2_001_trim_unpaired.fastq.gz /
ILLUMINACLIP:NexteraPE-PE.fa:2:30:10 /
MINLEN:40Now we can run the command for the other 3 pairs,
- WCCR_1913_S74_L002_R1_001 & WCCR_1913_S74_L002_R2_001
- WCCR_1913_S74_L003_R1_001 & WCCR_1913_S74_L003_R2_001
- WCCR_1913_S74_L004_R1_001 & WCCR_1913_S74_L004_R2_001
And then do the same for the rest of the samples.
Show/hide HYDRA TRIMMOMATIC job script
# /bin/sh
# ----------------Parameters---------------------- #
#$ -S /bin/sh
#$ -pe mthread 16
#$ -q sThC.q
#$ -cwd
#$ -j y
#$ -N job_00_trimmomatic
#$ -o hydra_logs/job_00_trimmomatic_redo.log
#
# ----------------Modules------------------------- #
module load bioinformatics/trimmomatic
#
# ----------------Your Commands------------------- #
#
echo + `date` job $JOB_NAME started in $QUEUE with jobID=$JOB_ID on $HOSTNAME
echo + NSLOTS = $NSLOTS
#
# ----------------COMMANDS------------------- #
#
echo = `date` job $JOB_NAME don
Snakemake Workflow
Thanks to the anvi’o implementation of Snakemake we can run many commands sequentially and/or simultaneously. There is plenty of good documentation on the anvi’o website about setting up Snakemake workflows here so we will refrain from any lengthy explanations.
You can see all the settings we used and download our configs file here or grab it from the folded code below. Some commands we chose not to run but they remain in the workflow for posterity.
Show/hide JSON-formatted configuration file.
{
"fasta_txt": "",
"workflow_name": "metagenomics",
"anvi_gen_contigs_database": {
"--project-name": "{group}",
"--description": "",
"--skip-gene-calling": "",
"--external-gene-calls": "",
"--ignore-internal-stop-codons": "",
"--skip-mindful-splitting": "",
"--contigs-fasta": "",
"--split-length": "",
"--kmer-size": "",
"--prodigal-translation-table": "",
"threads": ""
},
"centrifuge": {
"threads": 12,
"run": true,
"db": "/pool/genomics/stri_istmobiome/dbs/centrifuge_dbs/p+h+v"
},
"anvi_run_hmms": {
"run": true,
"threads": 5,
"--installed-hmm-profile": "",
"--hmm-profile-dir": ""
},
"anvi_run_ncbi_cogs": {
"run": true,
"threads": 5,
"--cog-data-dir": "/pool/genomics/stri_istmobiome/dbs/cog_db/",
"--sensitive": "",
"--temporary-dir-path": "/pool/genomics/stri_istmobiome/dbs/cog_db/tmp/",
"--search-with": ""
},
"anvi_run_scg_taxonomy": {
"run": true,
"threads": 6,
"--scgs-taxonomy-data-dir": "/pool/genomics/stri_istmobiome/dbs/scgs-taxonomy-data/"
},
"anvi_script_reformat_fasta": {
"run": true,
"--prefix": "{group}",
"--simplify-names": true,
"--keep-ids": "",
"--exclude-ids": "",
"--min-len": "1000",
"threads": ""
},
"emapper": {
"--database": "bact",
"--usemem": true,
"--override": true,
"path_to_emapper_dir": "",
"threads": ""
},
"anvi_script_run_eggnog_mapper": {
"--use-version": "0.12.6",
"run": "",
"--cog-data-dir": "/pool/genomics/stri_istmobiome/dbs/cog_db/",
"--drop-previous-annotations": "",
"threads": 20
},
"samples_txt": "samples.txt",
"metaspades": {
"additional_params": "--only-assembler",
"threads": 7,
"run": "",
"use_scaffolds": ""
},
"megahit": {
"--min-contig-len": 1000,
"--memory": 0.8,
"threads": 20,
"run": true,
"--min-count": "",
"--k-min": "",
"--k-max": "",
"--k-step": "",
"--k-list": "",
"--no-mercy": "",
"--no-bubble": "",
"--merge-level": "",
"--prune-level": "",
"--prune-depth": "",
"--low-local-ratio": "",
"--max-tip-len": "",
"--no-local": "",
"--kmin-1pass": "",
"--presets": "meta-sensitive",
"--mem-flag": "",
"--use-gpu": "",
"--gpu-mem": "",
"--keep-tmp-files": "",
"--tmp-dir": "",
"--continue": true,
"--verbose": ""
},
"idba_ud": {
"--min_contig": 1000,
"threads": 7,
"run": "",
"--mink": "",
"--maxk": "",
"--step": "",
"--inner_mink": "",
"--inner_step": "",
"--prefix": "",
"--min_count": "",
"--min_support": "",
"--seed_kmer": "",
"--similar": "",
"--max_mismatch": "",
"--min_pairs": "",
"--no_bubble": "",
"--no_local": "",
"--no_coverage": "",
"--no_correct": "",
"--pre_correction": ""
},
"iu_filter_quality_minoche": {
"run": true,
"--ignore-deflines": true,
"--visualize-quality-curves": "",
"--limit-num-pairs": "",
"--print-qual-scores": "",
"--store-read-fate": "",
"threads": ""
},
"gzip_fastqs": {
"run": true,
"threads": ""
},
"bowtie": {
"additional_params": "--no-unal",
"threads": 3
},
"samtools_view": {
"additional_params": "-F 4",
"threads": ""
},
"anvi_profile": {
"threads": 5,
"--sample-name": "{sample}",
"--overwrite-output-destinations": true,
"--report-variability-full": "",
"--skip-SNV-profiling": "",
"--profile-SCVs": truw,
"--description": "",
"--skip-hierarchical-clustering": "",
"--distance": "",
"--linkage": "",
"--min-contig-length": "",
"--min-mean-coverage": "",
"--min-coverage-for-variability": "",
"--cluster-contigs": "",
"--contigs-of-interest": "",
"--queue-size": "",
"--write-buffer-size": 10000,
"--max-contig-length": "",
"--max-coverage-depth": "",
"--ignore-orphans": ""
},
"anvi_merge": {
"--sample-name": "{group}",
"--overwrite-output-destinations": true,
"--description": "",
"--skip-hierarchical-clustering": "",
"--enforce-hierarchical-clustering": "",
"--distance": "",
"--linkage": "",
"threads": ""
},
"import_percent_of_reads_mapped": {
"run": true,
"threads": ""
},
"krakenuniq": {
"threads": 3,
"--gzip-compressed": true,
"additional_params": "",
"run": "",
"--db": "/scratch/genomics/scottjj/kraken_dbs/DB/"
},
"remove_short_reads_based_on_references": {
"delimiter-for-iu-remove-ids-from-fastq": " ",
"dont_remove_just_map": "",
"references_for_removal_txt": "",
"threads": ""
},
"anvi_cluster_contigs": {
"--collection-name": "{driver}",
"run": "",
"--driver": "concoct",
"--just-do-it": "",
"--additional-params-concoct": "",
"--additional-params-metabat2": "",
"--additional-params-maxbin2": "",
"--additional-params-dastool": "",
"--additional-params-binsanity": "",
"threads": 10
},
"gen_external_genome_file": {
"threads": ""
},
"export_gene_calls_for_centrifuge": {
"threads": ""
},
"anvi_import_taxonomy_for_genes": {
"threads": ""
},
"annotate_contigs_database": {
"threads": ""
},
"anvi_get_sequences_for_gene_calls": {
"threads": ""
},
"gunzip_fasta": {
"threads": ""
},
"reformat_external_gene_calls_table": {
"threads": ""
},
"reformat_external_functions": {
"threads": ""
},
"import_external_functions": {
"threads": ""
},
"anvi_run_pfams": {
"run": true,
"--pfam-data-dir": "/pool/genomics/stri_istmobiome/dbs/pfam_db",
"threads": 5
},
"iu_gen_configs": {
"--r1-prefix": "",
"--r2-prefix": "",
"threads": ""
},
"gen_qc_report": {
"threads": ""
},
"merge_fastqs_for_co_assembly": {
"threads": ""
},
"merge_fastas_for_co_assembly": {
"threads": ""
},
"bowtie_build": {
"threads": ""
},
"anvi_init_bam": {
"threads": ""
},
"krakenuniq_mpa_report": {
"threads": ""
},
"import_krakenuniq_taxonomy": {
"--min-abundance": "",
"threads": ""
},
"anvi_summarize": {
"additional_params": "",
"run": "",
"threads": ""
},
"anvi_split": {
"additional_params": "",
"run": "",
"threads": ""
},
"references_mode": "",
"all_against_all": "",
"kraken_txt": "",
"collections_txt": "",
"output_dirs": {
"FASTA_DIR": "02_FASTA",
"CONTIGS_DIR": "03_CONTIGS",
"QC_DIR": "01_QC",
"MAPPING_DIR": "04_MAPPING",
"PROFILE_DIR": "05_ANVIO_PROFILE",
"MERGE_DIR": "06_MERGED",
"TAXONOMY_DIR": "07_TAXONOMY",
"SUMMARY_DIR": "08_SUMMARY",
"SPLIT_PROFILES_DIR": "09_SPLIT_PROFILES",
"LOGS_DIR": "00_LOGS"
},
"max_threads": "",
"config_version": "1"
}
Since we are doing a co-assembly, we need a separate file called samples.txt, which can be downloaded here. This file tells anvi’o where to find the trimmed fastq files for each sample and what group the sample belongs to. The file is a four-column, tab-delimited file.
| sample | group | r1 | r2 |
|---|---|---|---|
| WCCR_1913 | WATER | comma separated list of forward reads | comma separated list of reverse reads |
| WCCR_1916 | WATER | comma separated list of forward reads | comma separated list of reverse reads |
| WROL_1914 | WATER | comma separated list of forward reads | comma separated list of reverse reads |
| WROL_1915 | WATER | comma separated list of forward reads | comma separated list of reverse reads |
Remember, there are 4 fastq files per direction (forward or reverse) per sample. So each comma separated list in the r1 column needs four file names and the same with the r2 column. And you must include relative path names.
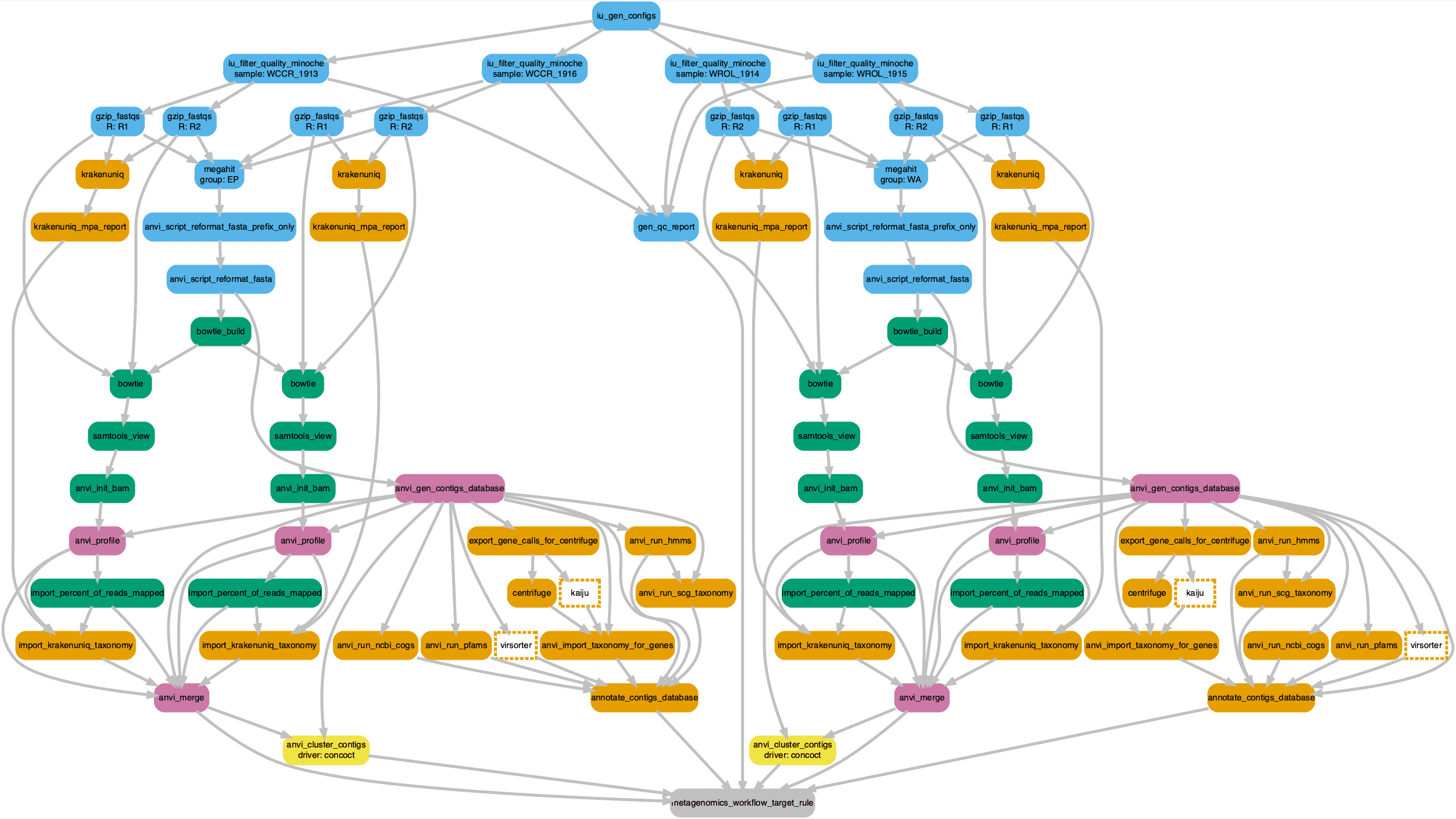
To see what will happen with the config file we can visualize the Snakemake workflow using a Directed acyclic graph (DAG) where edge connections represent dependencies and nodes represent commands. The workflow begins with trimmed reads and continues up to and including automatic binning of contigs. At the end of this workflow we then proceed with manual binning and MAG generation. We added nodes for VirSorter and Kaiju annotations since these are not part of the normal anvi’o workflow.
Click on the image to zoom in or download a copy.
Show/hide DAG R Code script
```r
dag <- grViz ("
digraph boxes_and_circles {
graph [layout = dot, align=center]
node [shape = rectangle, style = 'rounded,filled' fontname=sans, fontsize=12, penwidth=4]
edge[penwidth=4, color=grey];
0[label = 'metagenomics_workflow_target_rule', color = 'grey'];
1[label = 'anvi_merge', color = '#CC79A7'];
2[label = 'anvi_merge', color = '#CC79A7'];
3[label = 'annotate_contigs_database', color = '#E69F00'];
4[label = 'annotate_contigs_database', color = '#E69F00'];
5[label = 'gen_qc_report', color = '#56B4E9'];
6[label = 'anvi_cluster_contigs\ndriver: concoct', color = '#F0E442'];
7[label = 'anvi_cluster_contigs\ndriver: concoct', color = '#F0E442'];
8[label = 'anvi_gen_contigs_database', color = '#CC79A7'];
9[label = 'anvi_profile', color = '#CC79A7'];
10[label = 'anvi_profile', color = '#CC79A7'];
11[label = 'import_percent_of_reads_mapped', color = '#009E73'];
12[label = 'import_percent_of_reads_mapped', color = '#009E73'];
13[label = 'import_krakenuniq_taxonomy', color = '#E69F00'];
14[label = 'import_krakenuniq_taxonomy', color = '#E69F00'];
15[label = 'anvi_gen_contigs_database', color = '#CC79A7'];
16[label = 'anvi_profile', color = '#CC79A7'];
17[label = 'anvi_profile', color = '#CC79A7'];
18[label = 'import_percent_of_reads_mapped', color = '#009E73'];
19[label = 'import_percent_of_reads_mapped', color = '#009E73'];
20[label = 'import_krakenuniq_taxonomy', color = '#E69F00'];
21[label = 'import_krakenuniq_taxonomy', color = '#E69F00'];
22[label = 'anvi_import_taxonomy_for_genes', color = '#E69F00'];
23[label = 'anvi_run_hmms', color = '#E69F00'];
24[label = 'anvi_run_ncbi_cogs', color = '#E69F00'];
25[label = 'anvi_run_scg_taxonomy', color = '#E69F00'];
26[label = 'anvi_run_pfams', color = '#E69F00'];
27[label = 'anvi_import_taxonomy_for_genes', color = '#E69F00'];
28[label = 'anvi_run_hmms', color = '#E69F00'];
29[label = 'anvi_run_ncbi_cogs', color = '#E69F00'];
30[label = 'anvi_run_scg_taxonomy', color = '#E69F00'];
31[label = 'anvi_run_pfams', color = '#E69F00'];
32[label = 'iu_filter_quality_minoche\nsample: WCCR_1913', color = '#56B4E9'];
33[label = 'iu_filter_quality_minoche\nsample: WCCR_1916', color = '#56B4E9'];
34[label = 'iu_filter_quality_minoche\nsample: WROL_1914', color = '#56B4E9'];
35[label = 'iu_filter_quality_minoche\nsample: WROL_1915', color = '#56B4E9'];
36[label = 'anvi_script_reformat_fasta', color = '#56B4E9'];
37[label = 'anvi_init_bam', color = '#009E73'];
38[label = 'anvi_init_bam', color = '#009E73'];
39[label = 'krakenuniq_mpa_report', color = '#E69F00'];
40[label = 'krakenuniq_mpa_report', color = '#E69F00'];
41[label = 'anvi_script_reformat_fasta', color = '#56B4E9'];
42[label = 'anvi_init_bam', color = '#009E73'];
43[label = 'anvi_init_bam', color = '#009E73'];
44[label = 'krakenuniq_mpa_report', color = '#E69F00'];
45[label = 'krakenuniq_mpa_report', color = '#E69F00'];
46[label = 'centrifuge', color = '#E69F00'];
47[label = 'centrifuge', color = '#E69F00'];
48[label = 'iu_gen_configs', color = '#56B4E9'];
49[label = 'anvi_script_reformat_fasta_prefix_only', color = '#56B4E9'];
50[label = 'samtools_view', color = '#009E73'];
51[label = 'samtools_view', color = '#009E73'];
52[label = 'krakenuniq', color = '#E69F00'];
53[label = 'krakenuniq', color = '#E69F00'];
54[label = 'anvi_script_reformat_fasta_prefix_only', color = '#56B4E9'];
55[label = 'samtools_view', color = '#009E73'];
56[label = 'samtools_view', color = '#009E73'];
57[label = 'krakenuniq', color = '#E69F00'];
58[label = 'krakenuniq', color = '#E69F00'];
59[label = 'export_gene_calls_for_centrifuge', color = '#E69F00'];
60[label = 'export_gene_calls_for_centrifuge', color = '#E69F00'];
61[label = 'megahit\ngroup: EP', color = '#56B4E9'];
62[label = 'bowtie', color = '#009E73'];
63[label = 'bowtie', color = '#009E73'];
64[label = 'gzip_fastqs\nR: R1', color = '#56B4E9'];
65[label = 'gzip_fastqs\nR: R2', color = '#56B4E9'];
66[label = 'gzip_fastqs\nR: R1', color = '#56B4E9'];
67[label = 'gzip_fastqs\nR: R2', color = '#56B4E9'];
68[label = 'megahit\ngroup: WA', color = '#56B4E9'];
69[label = 'bowtie', color = '#009E73'];
70[label = 'bowtie', color = '#009E73'];
71[label = 'gzip_fastqs\nR: R1', color = '#56B4E9'];
72[label = 'gzip_fastqs\nR: R2', color = '#56B4E9'];
73[label = 'gzip_fastqs\nR: R1', color = '#56B4E9'];
74[label = 'gzip_fastqs\nR: R2', color = '#56B4E9'];
75[label = 'bowtie_build', color = '#009E73'];
76[label = 'bowtie_build', color = '#009E73'];
77[label = 'virsorter', color = '#E69F00', style = 'dashed'];
78[label = 'virsorter', color = '#E69F00', style = 'dashed'];
79[label = 'kaiju', color = '#E69F00', style = 'dashed'];
80[label = 'kaiju', color = '#E69F00', style = 'dashed'];
1->0; 2->0; 3->0; 4->0; 5->0; 6->0; 7->0; 8->1; 9->1;
10->1; 11->1; 12->1; 13->1; 14->1; 15->2; 16->2; 17->2;
18->2; 19->2; 20->2; 21->2; 8->3; 22->3; 23->3; 24->3;
25->3; 26->3; 15->4; 27->4; 28->4; 29->4; 30->4; 31->4;
{32 33 34 35}->5;
48->{32 33 34 35};
32->{64 65};
33->{66 67};
{64 65 66 67}->61;
{64 65}->52;
{66 67}->53;
34->{71 72};
35->{73 74};
{71 72 73 74}->68
{71 72}->57;
{73 74}->58;
8->6; 1->6; 15->7; 2->7;
36->8; 37->9; 8->9; 38->10; 8->10; 9->11; 10->12; 39->13;
11->13; 9->13; 40->14; 12->14; 10->14; 41->15; 42->16; 15->16;
43->17; 15->17; 16->18; 17->19; 44->20; 18->20; 16->20; 45->21;
19->21; 17->21; 46->22; 8->22; 8->23; 8->24; 23->25; 8->25;
8->26; 47->27; 15->27; 15->28; 15->29; 28->30; 15->30; 15->31;
49->36; 50->37; 51->38; 52->39;
53->40; 54->41; 55->42; 56->43; 57->44; 58->45; 59->46; 60->47;
61->49; 62->50; 63->51; 68->54;
69->55; 70->56; 8->59; 15->60;
75->62; 64->62; 65->62; 75->63;
66->63; 67->63; 76->69; 71->69; 72->69; 76->70; 73->70; 74->70;
36->75; 41->76; 79->22; 80->27;
15->77; 8->78; 77->4; 78->3; 59->79; 60->80;
graph [nodesep = 0.1]
{ rank=same; 13, 14, 20, 21 }
{ rank=same; 39, 40, 44, 45 }
{ rank=same; 32, 33, 34, 35 }
{ rank=same; 64, 65, 66, 67, 71, 72, 73, 74 }
{ rank=same; 5, 49, 54 }
}
")
export_svg(dag) %>%
charToRaw() %>%
rsvg() %>%
png::writePNG("images/dag.png")
```
And here are the commands we used to run the workflow.
anvi-run-workflow -w metagenomics -c default-config.txt --additional-params --jobs 63 --resources nodes=63 --keep-going --rerun-incomplete --unlock
anvi-run-workflow -w metagenomics -c default-config.txt --additional-params --jobs 63 --resources nodes=63 --keep-going --rerun-incompleteIf a Snakemake job fails the default behavior is to lock the workflow and because large jobs can fail for a lot of reasons, we decided to always include an identical command first with the --unlock flag. This will unlock the workflow and then the second command will execute. You can find an explanation of this here.
Show/hide HYDRA SNAKEMAKE job script
# /bin/sh
# ----------------Parameters---------------------- #
#$ -S /bin/sh
#$ -pe mthread 20
#$ -q sThM.q
#$ -l mres=140G,h_data=7G,h_vmem=7G,himem
#$ -cwd
#$ -j y
#$ -N job_01_run_mg_workflow
#$ -o hydra_logs/job_01_run_mg_workflow.log
#
# ----------------Modules------------------------- #
module load gcc/4.9.2
#
# ----------------Your Commands------------------- #
#
echo + `date` job $JOB_NAME started in $QUEUE with jobID=$JOB_ID on $HOSTNAME
echo + NSLOTS = $NSLOTS
#
# ----------------CALLING ANVIO------------------- #
export PATH=/home/scottjj/miniconda3:$PATH
export PATH=/home/scottjj/miniconda3/bin:$PATH
source activate anvio-master
#
# ----------------COOLIO?------------------- #
which python
python --version
source /home/scottjj/virtual-envs/anvio-master/bin/activate
which python
python --version
which anvi-interactive
diamond --version
anvi-self-test -v
#
# ----------------TEMP DIRECTORIES------------------- #
rm -r /pool/genomics/stri_istmobiome/dbs/cog_db/tmp/
mkdir -p /pool/genomics/stri_istmobiome/dbs/cog_db/tmp/
#
rm -r /pool/genomics/stri_istmobiome/dbs/pfam_db/tmp_data/
mkdir -p /pool/genomics/stri_istmobiome/dbs/pfam_db/tmp_data/
TMPDIR="/pool/genomics/stri_istmobiome/dbs/pfam_db/tmp_data/"
#
# ----------------COMMANDS------------------- #
#
anvi-run-workflow -w metagenomics -c default-config.txt --additional-params --jobs 20 --resources nodes=20 --keep-going --rerun-incomplete --unlock
anvi-run-workflow -w metagenomics -c default-config.txt --additional-params --jobs 20 --resources nodes=20 --keep-going --rerun-incomplete
echo = `date` job $JOB_NAME don
Snakemake Citations
There are many tools used in the workflow that need to be cited.
First there is the workflow engine itself—Snakemake (Köster and Rahmann 2012). QUALITY-FILTERING of raw reads was performed using Illumina Utils (Eren et al. 2013) implementation of the filter developed by Minoche and collegues (Minoche, Dohm, and Himmelbauer 2011). CO-ASSEMBLY of metagenomic samples performed using MEGAHIT (D. Li et al. 2015). GENE CALLING was performed using PRODIGAL (Hyatt et al. 2010). RECRUITMENT (a.k.a. mapping) of reads to assembly performed using BOWTIE2 (Langmead and Salzberg 2012) and SAMtools (H. Li et al. 2009). TAXONOMIC CLASSIFICATION of genes using CENTRIFUGE (Kim et al. 2016).
Also, there are a few tools that we ran outside of the Snakemake workflow. Results from these steps need to be added to the individual PROFILE.db’s, merged PROFILE.db, or CONTIGS.db. Therefore, before the anvi-merge portion of the Snakemake workflow finished, we killed the job, ran the accessory analyses described below, and then restarted the workflow to finish the missing step. Cumbersome, yes, but it got the job done.
Taxonomic Annotations
In this section we conduct taxonomic classification of short reads, contigs, and gene calls.
Kaken Annotation
In this section we use KrakenUniq (Breitwieser, Baker, and Salzberg 2018) to classify the short reads. Because of the memory demands of KrakenUniq, we could not get this to work in the Snakemake workflow, so we ran the analysis separately. For several of these commands we used lists of sample names and for loops to run through each sample.
Ok, here we run KrakenUniq and several reporting and summary steps against the short-read data for each sample using the list.txt file.
for sample in `cat list.txt`
do
krakenuniq --report-file $KRAKEN/$sample-REPORT.tsv 01_QC/$sample-QUALITY_PASSED_R1.fastq.gz 01_QC/$sample-QUALITY_PASSED_R2.fastq.gz --db $K_FILES/DB/ --threads $NSLOTS --preload --fastq-input --gzip-compressed --paired --output $KRAKEN/$sample-kraken.out
krakenuniq-report --db $K_FILES/DB/ $KRAKEN/$sample-kraken.out > $KRAKEN/$sample-kraken_report.txt
krakenuniq-mpa-report --header-line --db $K_FILES/DB/ $KRAKEN/$sample-kraken.out > $KRAKEN/$sample-kraken_mpa_report.txt
krakenuniq-translate --db $K_FILES/DB/ $KRAKEN/$sample-kraken.out > $KRAKEN/$sample-kraken.trans
doneNext, we format the output to make Krona plots.
for sample in `cat list.txt`
do
$KRA_to_KRON/kraken_to_krona.py $KRAKEN/$sample-kraken.trans > $KRAKEN/$sample-kraken.krona
doneAnd finally construct Krona (Ondov, Bergman, and Phillippy 2011) plots. We will come back to Krona plots when we explain what they are and how to use them in the Assembly & Annotation Summary section of the workflow.
for sample in `cat list.txt`
do
ktImportText -o $KRAKEN/$sample-kraken.html $KRAKEN/$sample-kraken.krona
doneWe can also make standalone HTML pages from each assembly containing all Krona plots.
ktImportText -o $KRAKEN/WATER-kraken.html $KRAKEN/WCCR_1913-kraken.krona $KRAKEN/WCCR_1916-kraken.krona $KRAKEN/WROL_1914-kraken.krona $KRAKEN/WROL_1915-kraken.kronaOr add the -c flag to make a single plot where samples are combined.
Now we have a Krona plot page for the water sample.
Show/hide HYDRA Kraken classification job details
# /bin/sh
# ----------------Parameters---------------------- #
#$ -S /bin/sh
#$ -pe mthread 3
#$ -q mThM.q
#$ -l mres=150G,h_data=150G,h_vmem=150G,himem
#$ -cwd
#$ -j y
#$ -N job_04_taxonomic_classification_kraken_reads
#$ -o hydra_logs/job_04_taxonomic_classification_kraken_reads.job
#
# ----------------Modules------------------------- #
#
# ----------------Load Envs------------------- #
#
echo + `date` job $JOB_NAME started in $QUEUE with jobID=$JOB_ID on $HOSTNAME
echo + NSLOTS = $NSLOTS
#
# ----------------Activate Kraken -------------- #
#
export PATH=/home/scottjj/miniconda3/bin:$PATH
source activate kraken
# ----------------SETUP Kraken Directories-------------- #
#
mkdir 07_TAXONOMY/KRAKEN/
mkdir 07_TAXONOMY/KRAKEN/READS
#
KRAKEN='/pool/genomics/stri_istmobiome/data/HYPOXIA_DATA/HYPOXIA/07_TAXONOMY/KRAKEN_TAXONOMY/MICROBIAL-NT/'
K_FILES='/scratch/genomics/scottjj/kraken_dbs/microbial-nt/'
KRA_to_KRON='/home/scottjj/miniconda3/envs/metawrap/bin/metawrap-scripts/'
# ----------------Run Kraken------------------- #
#
for sample in `cat list.txt`
do
krakenuniq --report-file $KRAKEN/$sample-REPORT.tsv 01_QC/$sample-QUALITY_PASSED_R1.fastq.gz 01_QC/$sample-QUALITY_PASSED_R2.fastq.gz --db $K_FILES/DB/ --threads $NSLOTS --preload --fastq-input --gzip-compressed --paired --output $KRAKEN/$sample-kraken.out
krakenuniq-report --db $K_FILES/DB/ $KRAKEN/$sample-kraken.out > $KRAKEN/$sample-kraken_report.txt
krakenuniq-mpa-report --header-line --db $K_FILES/DB/ $KRAKEN/$sample-kraken.out > $KRAKEN/$sample-kraken_mpa_report.txt
krakenuniq-translate --db $K_FILES/DB/ $KRAKEN/$sample-kraken.out > $KRAKEN/$sample-kraken.trans
done
source deactivate
#
source deactivate
#
# ----------------FORMAT FOR KRONA------------------- #
#
source activate metawrap
#
for sample in `cat list.txt`
do
$KRA_to_KRON/kraken_to_krona.py $KRAKEN/$sample-kraken.trans > $KRAKEN/$sample-kraken.krona
done
source deactivate
#
# ----------------MAKE KRONA PLOTS------------------- #
#
source activate krona
#
for sample in `cat list.txt`
#
do
ktImportText -o $KRAKEN/$sample-kraken.html $KRAKEN/$sample-kraken.krona
done
#
ktImportText -o $KRAKEN/WATER-kraken.html $KRAKEN/WCCR_1913-kraken.krona $KRAKEN/WCCR_1916-kraken.krona $KRAKEN/WROL_1914-kraken.krona $KRAKEN/WROL_1915-kraken.krona
#
source deactivate
#
echo = `date` job $JOB_NAME don
Kraken import
At this point we now add the kraken summary annotations to each individual PROFILE.db created in the Snakemake workflow. Again we used lists of sample names and for loops to run through each sample.
for sample in `cat list.txt`
do
anvi-import-taxonomy-for-layers -p 05_ANVIO_PROFILE/WATER/$sample/PROFILE.db --parse krakenuniq -i 07_TAXONOMY/KRAKEN_TAXONOMY/MICROBIAL-NT/$sample-kraken_mpa_report.txt
doneShow/hide HYDRA Import Kraken taxonomy job details
# /bin/sh
# ----------------Parameters---------------------- #
#$ -S /bin/sh
#$ -q sThC.q
#$ -l mres=5G,h_data=5G,h_vmem=5G
#$ -cwd
#$ -j y
#$ -N job_06_import_taxonomy_for_layers
#$ -o hydra_logs/job_06_import_taxonomy_for_layers.log
#
# ----------------Modules------------------------- #
module load gcc/4.9.2
#
# ----------------Your Commands------------------- #
#
echo + `date` job $JOB_NAME started in $QUEUE with jobID=$JOB_ID on $HOSTNAME
echo + NSLOTS = $NSLOTS
#
# ----------------Activate Anvio -------------- #
#
export PATH=/home/scottjj/miniconda3:$PATH
export PATH=/home/scottjj/miniconda3/bin:$PATH
source activate anvio-master
#
which python
python --version
source /home/scottjj/virtual-envs/anvio-master/bin/activate
which python
python --version
which anvi-interactive
diamond --version
anvi-self-test -v
#
# ----------------Import Kraken Annotations------------------- #
#
for sample in `cat list.txt`
#
do
anvi-import-taxonomy-for-layers -p 05_ANVIO_PROFILE/WATER/$sample/PROFILE.db --parse krakenuniq -i 07_TAXONOMY/KRAKEN_TAXONOMY/MICROBIAL-NT/$sample-kraken_mpa_report.txt
done
#
source deactivate
#
echo = `date` job $JOB_NAME don
After this you need to rerun anvi-merge so that the short-read annotations make it into the merged PROFILE.db. But we will get to that in a minute.
VirSorter Annotation
To classify any viral sequences, we ran VirSorter (Roux et al. 2015) on contigs from the co-assembly using our newly created contig.db generated after the co-assembly step. We ran VirSorter with and without the --virome flag. See issue 40 on the VirSorter GitHub site for a discussion of why we did this. We used DIAMOND (Buchfink, Xie, and Huson 2015) as the local sequence aligner.
Here is the VirSorter workflow.
wrapper_phage_contigs_sorter_iPlant.pl -f 02_FASTA/WATER/WATER-contigs.fa --ncpu $NSLOTS --db 2 --wdir 07_TAXONOMY/VIRSORTER/WATER --data-dir /pool/genomics/stri_istmobiome/dbs/virsorter/virsorter-data --diamond
wrapper_phage_contigs_sorter_iPlant.pl -f 02_FASTA/WATER/WATER-contigs.fa --ncpu $NSLOTS --db 2 --wdir 07_TAXONOMY/VIRSORTER_VIROME/WATER --data-dir /pool/genomics/stri_istmobiome/dbs/virsorter/virsorter-data --diamond --viromeShow/hide HYDRA VIRSORTER job script
# /bin/sh
# ----------------Parameters---------------------- #
#$ -S /bin/sh
#$ -pe mthread 10
#$ -q sThC.q
#$ -l mres=50G,h_data=5G,h_vmem=5G
#$ -cwd
#$ -j y
#$ -N job_03_run_virsorter
#$ -o hydra_logs/job_03_run_virsorter.log
#
# ----------------Modules------------------------- #
module load gcc/4.9.2
#
# ----------------Your Commands------------------- #
#
echo + `date` job $JOB_NAME started in $QUEUE with jobID=$JOB_ID on $HOSTNAME
echo + NSLOTS = $NSLOTS
#
export PATH=/home/scottjj/miniconda3:$PATH
export PATH=/home/scottjj/miniconda3/bin:$PATH
# ----------------Calling Virsorter------------------- #
#
source activate virsorter
which perl
#
# ----------------Make directories------------------- #
mkdir 07_TAXONOMY/
mkdir 07_TAXONOMY/VIRSORTER/
#
# ----------------COMMANDS------------------- #
wrapper_phage_contigs_sorter_iPlant.pl -f 02_FASTA/WATER/WATER-contigs.fa --ncpu $NSLOTS --db 2 --wdir 07_TAXONOMY/VIRSORTER/WATER --data-dir /pool/genomics/stri_istmobiome/dbs/virsorter/virsorter-data --diamond
#
wrapper_phage_contigs_sorter_iPlant.pl -f 02_FASTA/WATER/WATER-contigs.fa --ncpu $NSLOTS --db 2 --wdir 07_TAXONOMY/VIRSORTER_VIROME/WATER --data-dir /pool/genomics/stri_istmobiome/dbs/virsorter/virsorter-data --diamond --virome
#
source deactivate
#
echo = `date` job $JOB_NAME don
Kaiju Annotation
In addition to the Centrifuge classification we are going to use Kaiju (Menzel, Ng, and Krogh 2016) to classify gene calls. We do this against the two databases we built on the Annotation Databases page.
First we need to activate anvi’o, make a directory for the output, and grab the gene calls. Note that $KAIJU is the path to the input/output directory, which in this case is the new directory, 07_TAXONOMY/KAIJU/.
mkdir 07_TAXONOMY/KAIJU/
anvi-get-sequences-for-gene-calls -c 03_CONTIGS/WATER-contigs.db -o $KAIJU/WATER_gene_calls.fnaNext we run the Kaiju commands against the nr_euk database and then the mar database. Note that $K_FILES is the path to the database and $KAIJU is the path to the input/output directory.
# against nr_euk db
kaiju -t $K_FILES/nr_db/nodes.dmp -f $K_FILES/nr_db/nr_euk/kaiju_db_nr_euk.fmi -i $KAIJU/WATER_gene_calls.fna -o $KAIJU/WATER_kaiju_nr.out -z 16 -v
# against mar db
kaiju -t $K_FILES/marine_db/nodes.dmp -f $K_FILES/marine_db/marine_db/kaiju_db_mar.fmi -i $KAIJU/WATER_gene_calls.fna -o $KAIJU/WATER_kaiju_mar.out -z 16 -vFinally, we need to add taxon names to the output in order to import the taxonomy into the contig.db later on. We will also compare the output of the two classifications.
# against nr_euk db
kaiju-addTaxonNames -t $K_FILES/nr_db/nodes.dmp -n $K_FILES/nr_db/names.dmp -i $KAIJU/WATER_kaiju_nr.out -o $KAIJU/WATER_kaiju_nr.names -r superkingdom,phylum,order,class,family,genus,species
# against mar db
kaiju-addTaxonNames -t $K_FILES/marine_db/nodes.dmp -n $K_FILES/marine_db/names.dmp -i $KAIJU/WATER_kaiju_mar.out -o $KAIJU/WATER_kaiju_mar.names -r superkingdom,phylum,order,class,family,genus,speciesShow/hide HYDRA KAIJU job script
# /bin/sh
# ----------------Parameters---------------------- #
#$ -S /bin/sh
#$ -pe mthread 20
#$ -q sThC.q
#$ -l mres=120G,h_data=6G,h_vmem=6G
#$ -cwd
#$ -j y
#$ -N job_04_taxonomic_classification_kaiju
#$ -o hydra_logs/job_04_taxonomic_classification_kaiju.job
#
# ----------------Modules------------------------- #
#
# ----------------Load Envs------------------- #
#
echo + `date` job $JOB_NAME started in $QUEUE with jobID=$JOB_ID on $HOSTNAME
echo + NSLOTS = $NSLOTS
#
export PATH=/home/scottjj/miniconda3:$PATH
export PATH=/home/scottjj/miniconda3/bin:$PATH
export PATH=/home/scottjj/miniconda3/envs/kaiju/bin:$PATH
export PATH=/home/scottjj/miniconda3/envs/krona/bin:$PATH
export PATH=/pool/genomics/stri_istmobiome/dbs/kaiju_db/:$PATH
#
#NOT SURE IF THE FOLLOWING TWO LINES ARE NEEDED
export PERL5LIB="/home/scottjj/miniconda3/envs/kaiju/lib/5.26.2"
export PERL5LIB="/home/scottjj/miniconda3/envs/kaiju/lib/5.26.2/x86_64-linux-thread-multi:$PERL5LIB"
# ----------------SETUP KAIJU Directories-------------- #
#
mkdir 07_TAXONOMY/KAIJU/
KAIJU='/pool/genomics/stri_istmobiome/data/HYPOXIA_DATA/HYPOXIA/07_TAXONOMY/KAIJU/'
K_FILES='/pool/genomics/stri_istmobiome/dbs/kaiju_db'
#
# ----------------Get Gene Files------------------- #
source activate anvio-6.1
#
anvi-get-sequences-for-gene-calls -c 03_CONTIGS/WATER-contigs.db -o $KAIJU/WATER_gene_calls.fna
#
source deactivate
#
# ----------------RUN KAIJU------------------- #
source activate kaiju
which kaiju
gcc --version
which perl
# ----------------AGAINST nr DB------------------- #
#
kaiju -t $K_FILES/nr_db/nodes.dmp -f $K_FILES/nr_db/nr_euk/kaiju_db_nr_euk.fmi -i $KAIJU/WATER_gene_calls.fna -o $KAIJU/WATER_kaiju_nr.out -z 16 -v
#
kaiju-addTaxonNames -t $K_FILES/nr_db/nodes.dmp -n $K_FILES/nr_db/names.dmp -i $KAIJU/WATER_kaiju_nr.out -o $KAIJU/WATER_kaiju_nr.names -r superkingdom,phylum,order,class,family,genus,species
#
# ----------------AGAINST marine DB------------------- #
#
kaiju -t $K_FILES/marine_db/nodes.dmp -f $K_FILES/marine_db/marine_db/kaiju_db_mar.fmi -i $KAIJU/WATER_gene_calls.fna -o $KAIJU/WATER_kaiju_mar.out -z 16 -v
#
kaiju-addTaxonNames -t $K_FILES/marine_db/nodes.dmp -n $K_FILES/marine_db/names.dmp -i $KAIJU/WATER_kaiju_mar.out -o $KAIJU/WATER_kaiju_mar.names -r superkingdom,phylum,order,class,family,genus,species
#
source deactivate
#
echo = `date` job $JOB_NAME don
Kaiju Import
Now it is time to add the Kaiju annotation to the contig.db. We will also use the opportunity to generate some Krona plots. We will discuss these in more detail in the next chapter.
First we need to take the Kaiju-formatted output file and format it from Krona. We can do this for the results from both annotations.
# from nr_euk db
kaiju2krona -t $K_FILES/nr_db/nodes.dmp -n $K_FILES/nr_db/names.dmp -i $KAIJU/WATER_kaiju_nr.out -o $KAIJU/WATER_kaiju_nr.out.krona
# from mar db
kaiju2krona -t $K_FILES/marine_db/nodes.dmp -n $K_FILES/marine_db/names.dmp -i $KAIJU/WATER_kaiju_mar.out -o $KAIJU/WATER_kaiju_mar.out.kronaWhile we are at it, lets also generate some summary files of taxonomic content, again from both annotations.
# from nr_euk db
kaiju2table -t $K_FILES/nr_db/nodes.dmp -n $K_FILES/nr_db/names.dmp -r class $KAIJU/WATER_kaiju_nr.out -l phylum,class,order,family -o $KAIJU/WATER_kaiju_nr.out.summary
# from mar db
kaiju2table -t $K_FILES/marine_db/nodes.dmp -n $K_FILES/marine_db/names.dmp -r class $KAIJU/WATER_kaiju_mar.out -l phylum,class,order,family -o $KAIJU/WATER_kaiju_mar.out.summaryNow we will the output files from above to generate Krona plots for each annotation and send the output to HTML files. For this we use the krona conda package.
# from nr_euk db
ktImportText -o $KAIJU/WATER_kaiju_nr.out.html $KAIJU/WATER_kaiju_nr.out.krona
#from mar db
ktImportText -o $KAIJU/WATER_kaiju_mar.out.html $KAIJU/WATER_kaiju_mar.out.kronaFinally, it is time to import the Kaiju taxonomies into the contig.db for each assembly. As we will see later, the mar db annotations called very few viral sequences, even though we know from Virsorter there are a lot of viruses. Therefore, we decided to use the nr db annotations.
anvi-import-taxonomy-for-genes -c 03_CONTIGS/WATER-contigs.db -p kaiju -i $KAIJU/WATER_kaiju_nr.names --just-do-itShow/hide HYDRA KAIJU import and summarize job script
# /bin/sh
# ----------------Parameters---------------------- #
#$ -S /bin/sh
#$ -pe mthread 5
#$ -q sThC.q
#$ -l mres=25G,h_data=5G,h_vmem=5G
#$ -cwd
#$ -j y
#$ -N job_05_kaiju_summary
#$ -o hydra_logs/job_05_kaiju_summary.job
#
# ----------------Modules------------------------- #
#
# ----------------Load Envs------------------- #
#
echo + `date` job $JOB_NAME started in $QUEUE with jobID=$JOB_ID on $HOSTNAME
echo + NSLOTS = $NSLOTS
#
export PATH=/home/scottjj/miniconda3:$PATH
export PATH=/home/scottjj/miniconda3/bin:$PATH
export PATH=/home/scottjj/miniconda3/envs/kaiju/bin:$PATH
export PATH=/home/scottjj/miniconda3/envs/krona/bin:$PATH
export PATH=/pool/genomics/stri_istmobiome/dbs/kaiju_db/:$PATH
#
#NOT SURE IF THE FOLLOWING TWO LINES ARE NEEDED
export PERL5LIB="/home/scottjj/miniconda3/envs/kaiju/lib/5.26.2"
export PERL5LIB="/home/scottjj/miniconda3/envs/kaiju/lib/5.26.2/x86_64-linux-thread-multi:$PERL5LIB"
# ----------------SETUP KAIJU Directories-------------- #
#
KAIJU='/pool/genomics/stri_istmobiome/data/HYPOXIA_DATA/HYPOXIA/07_TAXONOMY/KAIJU/'
K_FILES='/pool/genomics/stri_istmobiome/dbs/kaiju_db/'
#
# ----------------Activate------------------- #
source activate kaiju
which kaiju
gcc --version
which perl
# ----------------get files for KRONA plots------------------- #
#
# ----------------nr_euk db------------------- #
kaiju2krona -t $K_FILES/nr_db/nodes.dmp -n $K_FILES/nr_db/names.dmp -i $KAIJU/WATER_kaiju_nr.out -o $KAIJU/WATER_kaiju_nr.out.krona
#
kaiju2table -t $K_FILES/nr_db/nodes.dmp -n $K_FILES/nr_db/names.dmp -r class $KAIJU/WATER_kaiju_nr.out -l phylum,class,order,family -o $KAIJU/WATER_kaiju_nr.out.summary
#
# ----------------mar db------------------- #
#
kaiju2krona -t $K_FILES/marine_db/nodes.dmp -n $K_FILES/marine_db/names.dmp -i $KAIJU/WATER_kaiju_mar.out -o $KAIJU/WATER_kaiju_mar.out.krona
#
kaiju2table -t $K_FILES/marine_db/nodes.dmp -n $K_FILES/marine_db/names.dmp -r class $KAIJU/WATER_kaiju_mar.out -l phylum,class,order,family -o $KAIJU/WATER_kaiju_mar.out.summary
#
# ----------------Build KRONA plots------------------- #
source activate krona
ktImportText -o $KAIJU/WATER_kaiju_nr.out.html $KAIJU/WATER_kaiju_nr.out.krona
#
ktImportText -o $KAIJU/WATER_kaiju_mar.out.html $KAIJU/WATER_kaiju_mar.out.krona
conda deactivate
#--------------------ANVIO PARSER--------------#
source activate anvio-master
#
which python
python --version
source /home/scottjj/virtual-envs/anvio-master/bin/activate
which python
python --version
which anvi-interactive
diamond --version
anvi-self-test -v
#
anvi-import-taxonomy-for-genes -c 03_CONTIGS/WATER-contigs.db -p kaiju -i $KAIJU/WATER_kaiju_nr.names --just-do-it
#
source deactivate
#
echo = `date` job $JOB_NAME don
Functional Annotations
SCG Taxonomy & Pfam
Here we run single-copy core genes in the contigs.db with taxonomic names against a local SCG taxonomy database. After this we can run anvi-estimate-scg-taxonomy to estimate taxonomy at genome-, collection-, or metagenome-level. We also needed to rerun the Pfam analysis because the original workflow failed at this step because of an improperly entered db path.
# Against SCG
anvi-run-scg-taxonomy -c 03_CONTIGS/WAATER-contigs.db -P 1 -T $NSLOTS
# Against Pfam
anvi-run-pfams -c 03_CONTIGS/WATER-contigs.db --pfam-data-dir $PATH/pfam_db/ -T $NSLOTSShow/hide HYDRA SCG & Pfam job details
# /bin/sh
# ----------------Parameters---------------------- #
#$ -S /bin/sh
#$ -pe mthread 20
#$ -q mThM.q
#$ -l mres=200G,h_data=10G,h_vmem=10G,himem
#$ -cwd
#$ -j y
#$ -N job_07_run_scg_tax_and_pfam
#$ -o hydra_logs/job_07_run_scg_tax_and_pfam.log
#
# ----------------Modules------------------------- #
module load gcc/4.9.2
#
# ----------------Your Commands------------------- #
#
echo + `date` job $JOB_NAME started in $QUEUE with jobID=$JOB_ID on $HOSTNAME
echo + NSLOTS = $NSLOTS
#
# ----------------Activate Anvio -------------- #
#
export PATH=/home/scottjj/miniconda3:$PATH
export PATH=/home/scottjj/miniconda3/bin:$PATH
source activate anvio-master
#
which python
python --version
source /home/scottjj/virtual-envs/anvio-master/bin/activate
which python
python --version
which anvi-interactive
diamond --version
anvi-self-test -v
#
# ----------------Setup tmp directories------------------- #
rm -r /pool/genomics/stri_istmobiome/dbs/scgs-taxonomy-data/tmp_data/
mkdir -p /pool/genomics/stri_istmobiome/dbs/scgs-taxonomy-data/tmp_data/
TMPDIR="/pool/genomics/stri_istmobiome/dbs/scgs-taxonomy-data/tmp_data/"
#
rm -r /pool/genomics/stri_istmobiome/dbs/pfam_db/tmp_data/
mkdir -p /pool/genomics/stri_istmobiome/dbs/pfam_db/tmp_data/
TMPDIR="/pool/genomics/stri_istmobiome/dbs/pfam_db/tmp_data/"
#
# ----------------run anvio commands------------------- #
#
anvi-run-scg-taxonomy -c 03_CONTIGS/WATER-contigs.db -P 1 -T $NSLOTS
#
anvi-run-pfams -c 03_CONTIGS/WAATER-contigs.db --pfam-data-dir /pool/genomics/stri_istmobiome/dbs/pfam_db/ -T $NSLOTS
#
echo = `date` job $JOB_NAME done
GhostKOALA Annotations
The last thing we can do is run KEGG (Kanehisa and Goto 2000) annotations using the GhostKOALA server (Kanehisa, Sato, and Morishima 2016). Running GhostKOALA/KEGG was pretty easy thanks to this handy tutorial.
The steps we used to annotate genes with GhostKOALA/KEGG are reproduced from the tutorial.
A little setup first. Make a directory for the analysis and grab the parsing script.
mkdir -p GhostKOALA/
git clone https://github.com/edgraham/GhostKoalaParser.gitNow we need to export anvio gene calls as amino acid sequences.
anvi-get-sequences-for-gene-calls -c 03_CONTIGS/WATER-contigs.db --get-aa-sequences -o GhostKOALA/WATER-protein-sequences.fa
anvi-get-sequences-for-gene-calls -c 03_CONTIGS/MAT-contigs.db --get-aa-sequences -o GhostKOALA/MAT-protein-sequences.faBecause of a peculiarity with the way the server handles fasta files we need to modify the fasta headers. Anvio gene calls begin with a digit and the server doesn’t like that. So, we will the prefix genecall to every gene call ID.
sed -i 's/>/>genecall_/' GhostKOALA/WATER-protein-sequences.fa
sed -i 's/>/>genecall_/' GhostKOALA/MAT-protein-sequences.faAt this point we can upload our modified amino acid gene call files to the GhostKOALA server.
You can only run one instance of GhostKOALA at a time per email address and the upload limit is 300Mb. You may need to split large fasta files into smaller chunks.
Show/hide HYDRA GhostKOALA annotation import job script
# /bin/sh
# ----------------Parameters---------------------- #
#$ -S /bin/sh
#$ -pe mthread 20
#$ -q sThM.q
#$ -l mres=140G,h_data=7G,h_vmem=7G,himem
#$ -cwd
#$ -j y
#$ -N job_06_run_ghostkoala
#$ -o hydra_logs/job_01_run_ghostkoala.log
#$ -M scottjj@si.edu
#
# ----------------Modules------------------------- #
module load gcc/4.9.2
#
# ----------------Your Commands------------------- #
#
echo + `date` job $JOB_NAME started in $QUEUE with jobID=$JOB_ID on $HOSTNAME
echo + NSLOTS = $NSLOTS
#
# ----------------Activate Anvio -------------- #
#
export PATH=/home/scottjj/miniconda3:$PATH
export PATH=/home/scottjj/miniconda3/bin:$PATH
source activate anvio-master
#
which python
python --version
source /home/scottjj/virtual-envs/anvio-master/bin/activate
which python
python --version
which anvi-interactive
diamond --version
anvi-self-test -v
#
# ----------------Setup -------------- #
#
mkdir -p GhostKOALA/
git clone https://github.com/edgraham/GhostKoalaParser.git
# ----------------Export Anvio Gene Calls -------------- #
#
anvi-get-sequences-for-gene-calls -c 03_CONTIGS/WATER-contigs.db --get-aa-sequences -o GhostKOALA/WATER-protein-sequences.fa
anvi-get-sequences-for-gene-calls -c 03_CONTIGS/MAT-contigs.db --get-aa-sequences -o GhostKOALA/MAT-protein-sequences.fa
# ----------------Reformat fata headers -------------- #
#
sed -i 's/>/>genecall_/' 07_GhostKOALA/WATER-protein-sequences.fa
sed -i 's/>/>genecall_/' 07_GhostKOALA/MAT-protein-sequences.fa
# ----------------STOP!!! Run GhostKOALA-------------- #
# At this point you must upload modified gene calls to the SERVER
# http://www.kegg.jp/ghostkoala/
#
echo = `date` job $JOB_NAME don
Import GhostKOALA
Once the jobs have finished we can import the data into our contig databases. In the repository you cloned earlier there is a file called KO_Orthology_ko00001.txt. See this section for an explanation of the next step about converting the KEGG Orthology assignments to functions. We need this htext file to match the orthologies with function.
mkdir GhostKOALA/GhostKoalaParser
wget 'https://www.genome.jp/kegg-bin/download_htext?htext=ko00001&format=htext&filedir=' -O GhostKOALA/GhostKoalaParser/ko00001.keg
# Set the variable path
kegfile="GhostKOALA/GhostKoalaParser/ko00001.keg"Now we need to parse the file using this code snippet from the tutorial.
while read -r prefix content
do
case "$prefix" in A) col1="$content";; \
B) col2="$content" ;; \
C) col3="$content";; \
D) echo -e "$col1\t$col2\t$col3\t$content";;
esac
done < <(sed '/^[#!+]/d;s/<[^>]*>//g;s/^./& /' < "$kegfile") > GhostKOALA/GhostKoalaParser/KO_Orthology_ko00001.txtTime to parse the annotation file…
python GhostKOALA/GhostKoalaParser/KEGG-to-anvio --KeggDB GhostKOALA/GhostKoalaParser//KO_Orthology_ko00001.txt -i GhostKOALA/water_ko.txt -o GhostKOALA/water-KeggAnnotations-AnviImportable.txt
python GhostKOALA/GhostKoalaParser/KEGG-to-anvio --KeggDB GhostKOALA/GhostKoalaParser//KO_Orthology_ko00001.txt -i GhostKOALA/mat_ko.txt -o GhostKOALA/mat-KeggAnnotations-AnviImportable.txt…and parse the KEGG taxonomy
python GhostKOALA/GhostKoalaParser/GhostKOALA-taxonomy-to-anvio GhostKOALA/water.out.top GhostKOALA/water-KeggTaxonomy.txt
python GhostKOALA/GhostKoalaParser/GhostKOALA-taxonomy-to-anvio GhostKOALA/mat.out.top GhostKOALA/mat-KeggTaxonomy.txtNow we can import it all into the anvio contig databases.
anvi-import-functions -c 03_CONTIGS/WATER-contigs.db -i GhostKOALA/water-KeggAnnotations-AnviImportable.txt
anvi-import-functions -c 03_CONTIGS/MAT-contigs.db -i GhostKOALA/mat-KeggAnnotations-AnviImportable.txt
#
anvi-import-taxonomy-for-genes -c 03_CONTIGS/WATER-contigs.db -i GhostKOALA/water-KeggTaxonomy.txt -p default_matrix
anvi-import-taxonomy-for-genes -c 03_CONTIGS/MAT-contigs.db -i GhostKOALA/mat-KeggTaxonomy.txt -p default_matrixAnd that’s it!
Show/hide HYDRA GhostKOALA parsing job script
# /bin/sh
# ----------------Parameters---------------------- #
#$ -S /bin/sh
#$ -pe mthread 20
#$ -q sThM.q
#$ -l mres=140G,h_data=7G,h_vmem=7G,himem
#$ -cwd
#$ -j y
#$ -N job_06_run_ghostkoala2
#$ -o hydra_logs/job_01_run_ghostkoala2.log
#$ -M scottjj@si.edu
#
# ----------------Modules------------------------- #
module load gcc/4.9.2
#
# ----------------Your Commands------------------- #
#
echo + `date` job $JOB_NAME started in $QUEUE with jobID=$JOB_ID on $HOSTNAME
echo + NSLOTS = $NSLOTS
#
# ----------------Activate Anvio -------------- #
#
export PATH=/home/scottjj/miniconda3:$PATH
export PATH=/home/scottjj/miniconda3/bin:$PATH
source activate anvio-master
#
which python
python --version
source /home/scottjj/virtual-envs/anvio-master/bin/activate
which python
python --version
which anvi-interactive
diamond --version
anvi-self-test -v
#
# ----------------Generate the KEGG orthology table-------------- #
mkdir GhostKOALA/GhostKoalaParser
wget 'https://www.genome.jp/kegg-bin/download_htext?htext=ko00001&format=htext&filedir=' -O GhostKOALA/GhostKoalaParser/ko00001.keg
#
kegfile="GhostKOALA/GhostKoalaParser/ko00001.keg"
#
while read -r prefix content
do
case "$prefix" in A) col1="$content";; \
B) col2="$content" ;; \
C) col3="$content";; \
D) echo -e "$col1\t$col2\t$col3\t$content";;
esac
done < <(sed '/^[#!+]/d;s/<[^>]*>//g;s/^./& /' < "$kegfile") > GhostKOALA/GhostKoalaParser/KO_Orthology_ko00001.txt
#
# ----------------Parsing the results from GhostKOALA-------------- #
#
python GhostKOALA/GhostKoalaParser/KEGG-to-anvio --KeggDB GhostKOALA/GhostKoalaParser//KO_Orthology_ko00001.txt -i GhostKOALA/water_ko.txt -o GhostKOALA/water-KeggAnnotations-AnviImportable.txt
python GhostKOALA/GhostKoalaParser/KEGG-to-anvio --KeggDB GhostKOALA/GhostKoalaParser//KO_Orthology_ko00001.txt -i GhostKOALA/mat_ko.txt -o GhostKOALA/mat-KeggAnnotations-AnviImportable.txt
#
python GhostKOALA/GhostKoalaParser/GhostKOALA-taxonomy-to-anvio GhostKOALA/water.out.top GhostKOALA/water-KeggTaxonomy.txt
python GhostKOALA/GhostKoalaParser/GhostKOALA-taxonomy-to-anvio GhostKOALA/mat.out.top GhostKOALA/mat-KeggTaxonomy.txt
# ----------------Importing GhostKOALA results-------------- #
#
anvi-import-functions -c 03_CONTIGS/WATER-contigs.db -i GhostKOALA/water-KeggAnnotations-AnviImportable.txt
anvi-import-functions -c 03_CONTIGS/MAT-contigs.db -i GhostKOALA/mat-KeggAnnotations-AnviImportable.txt
#
anvi-import-taxonomy-for-genes -c 03_CONTIGS/WATER-contigs.db -i GhostKOALA/water-KeggTaxonomy.txt -p default_matrix
anvi-import-taxonomy-for-genes -c 03_CONTIGS/MAT-contigs.db -i GhostKOALA/mat-KeggTaxonomy.txt -p default_matrix
#
echo = `date` job $JOB_NAME don
Merge Profiles & Import VirSorter
Now that everything is added into the contig.db for the EP and WA assemblies and layer taxonomy is added to the individual PROFILE.db, its now time to merge all PROFILE.db into a single database for each assembly.
anvi-merge 05_ANVIO_PROFILE/WATER/*/PROFILE.db -c 03_CONTIGS/WATER-contigs.db -o 06_MERGED/WATERShow/hide HYDRA Merge Profile databases job details
# /bin/sh
# ----------------Parameters---------------------- #
#$ -S /bin/sh
#$ -pe mthread 2
#$ -q mThM.q
#$ -l mres=20G,h_data=10G,h_vmem=10G,himem
#$ -cwd
#$ -j y
#$ -N job_08_merge_profile_wa
#$ -o hydra_logs/job_08_merge_profile_wa.log
#
# ----------------Modules------------------------- #
module load gcc/4.9.2
#
# ----------------Your Commands------------------- #
#
echo + `date` job $JOB_NAME started in $QUEUE with jobID=$JOB_ID on $HOSTNAME
echo + NSLOTS = $NSLOTS
#
# ----------------Activate Anvio -------------- #
#
export PATH=/home/scottjj/miniconda3:$PATH
export PATH=/home/scottjj/miniconda3/bin:$PATH
source activate anvio-master
#
which python
python --version
source /home/scottjj/virtual-envs/anvio-master/bin/activate
which python
python --version
which anvi-interactive
diamond --version
anvi-self-test -v
#
# ----------------run anvio commands------------------- #
#
anvi-merge 05_ANVIO_PROFILE/WATER/*/PROFILE.db -c 03_CONTIGS/WATER-contigs.db -o 06_MERGED/WATER/
#
echo = `date` job $JOB_NAME done
VirSorter Import
Now that we have a merged profile database for each assembly, we can deal with the VirSorter data. We did this last for two reasons. First, we wanted to get a better idea of the total abundance of the viral community by looking at the other taxonomic annotations. Since the viral community is > 10% we decided to use the virome decontamination data from VirSorter. Second, we need to add the VirSorter annotations to the merged PROFILE.dbs as a COLLECTION.
This is a multi-step process that is explained in great detail here. But please note that depending on the state of affairs with VirSorter and/or anvi’o, you may need to modify the output of anvi-export-gene-calls per VirSorter GitHub issue 65.
Show/hide the issue and the fix
The tool virsorter_to_anvio.py uses the output of anvi-export-gene-calls (among other files)
to import virsorter annotation into anvio dbs. The script relies on the output columns to be
in a particular order. That order has changed at some point in anvio's recent past.
#
VirSorter requires the following format:
gene_callers_id contig start stop direction partial source version
#
But it is now:
gene_callers_id contig direction partial source start stop version aa_sequence
#
So, the columns need to be rearranged or the script changed.
I wish I could offer I script fix :) but I just used awk.
#
awk 'BEGIN {FS="\t"; OFS="\t"} {print $1, $2, $6, $7, $3, $4, $5, $8}' all_gene_calls_TEMP.txt > all_gene_calls.txt
#
This rearranges and eliminates the aa_sequence column.
First we need to grab some parsing scripts.
wget https://raw.githubusercontent.com/brymerr921/VirSorterParser/master/virsorter_to_anvio.py -P $VIRSORTER/helper_scripts
wget https://raw.githubusercontent.com/brymerr921/VirSorterParser/master/hallmark_to_function_files/db1_hallmark_functions.txt -P $VIRSORTER/helper_scripts
wget https://raw.githubusercontent.com/brymerr921/VirSorterParser/master/hallmark_to_function_files/db2_hallmark_functions.txt -P $VIRSORTER/helper_scriptsNext, export the files we need for the annotations from the contig databases.
anvi-export-table 03_CONTIGS/WATER-contigs.db --table splits_basic_info -o $VIRSORTER/WATER-splits_basic_info.txt
#
anvi-export-gene-calls -c 03_CONTIGS/WATER-contigs.db -o $VIRSORTER/WATER-all_gene_calls.txtRun the VirSorter parsing scripts.
python $VIRSORTER/helper_scripts/virsorter_to_anvio.py --db 2 -a $VIRSORTER/WATER/Metric_files/VIRSorter_affi-contigs.tab -g $VIRSORTER/WATER/VIRSorter_global-phage-signal.csv -s $VIRSORTER/WATER-splits_basic_info.txt -n $VIRSORTER/WATER-all_gene_calls.txt -f $VIRSORTER/helper_scripts/db2_hallmark_functions.txt -A $VIRSORTER/WATER-virsorter_additional_info.txt -F $VIRSORTER/WATER-virsorter_annotations.txt -C $VIRSORTER/WATER-virsorter_collection.txt
#If you get an error you may need to reformat the gene calls files like so.
awk 'BEGIN {FS="\t"; OFS="\t"} {print $1, $2, $6, $7, $3, $4, $5, $8}' all_gene_calls_TEMP.txt > all_gene_calls.txtFinally, import all of the data in to the contig and profile dbs from each assembly.
anvi-import-misc-data $VIRSORTER/WATER-virsorter_additional_info.txt -p 06_MERGED/WATER/PROFILE.db --target-data-table items
anvi-import-collection $VIRSORTER/WATER-virsorter_collection.txt -c 03_CONTIGS/WATER-contigs.db -p 06_MERGED/WATER/PROFILE.db -C VIRSORTER
anvi-import-functions -c 03_CONTIGS/WATER-contigs.db -i $VIRSORTER/WATER-virsorter_annotations.txtShow/hide HYDRA Virsorter annotation import job details
# /bin/sh
# ----------------Parameters---------------------- #
#$ -S /bin/sh
#$ -pe mthread 10
#$ -q sThC.q
#$ -l mres=50G,h_data=5G,h_vmem=5G
#$ -cwd
#$ -j y
#$ -N job_09_parse_virsorter
#$ -o hydra_logs/job_09_parse_virsorter.log
#
# ----------------Modules------------------------- #
module load gcc/4.9.2
#
# ----------------Your Commands------------------- #
#
echo + `date` job $JOB_NAME started in $QUEUE with jobID=$JOB_ID on $HOSTNAME
echo + NSLOTS = $NSLOTS
#
# ----------------Activate Anvio -------------- #
#
export PATH=/home/scottjj/miniconda3:$PATH
export PATH=/home/scottjj/miniconda3/bin:$PATH
source activate anvio-master
#
which python
python --version
source /home/scottjj/virtual-envs/anvio-master/bin/activate
which python
python --version
which anvi-interactive
diamond --version
anvi-self-test -v
#
# ----------------Get VirSorter Scripts------------------- #
#
VIRSORTER='/pool/genomics/stri_istmobiome/data/HYPOXIA_DATA/HYPOXIA/07_TAXONOMY/VIRSORTER_VIROME'
#
wget https://raw.githubusercontent.com/brymerr921/VirSorterParser/master/virsorter_to_anvio.py -P $VIRSORTER/helper_scripts
wget https://raw.githubusercontent.com/brymerr921/VirSorterParser/master/hallmark_to_function_files/db1_hallmark_functions.txt -P $VIRSORTER/helper_scripts
wget https://raw.githubusercontent.com/brymerr921/VirSorterParser/master/hallmark_to_function_files/db2_hallmark_functions.txt -P $VIRSORTER/helper_scripts
#
# ----------------Export files for annotation------------------- #
#
anvi-export-table 03_CONTIGS/WATER-contigs.db --table splits_basic_info -o $VIRSORTER/WATER-splits_basic_info.txt
anvi-export-gene-calls -c 03_CONTIGS/WATER-contigs.db -o $VIRSORTER/WATER-all_gene_calls_TEMP.txt --gene-caller prodigal
#
# ----------------REORDER columns !!!!!!!!MAY NOT NEED TO DO THIS!!!!!------------------- #
#
awk 'BEGIN {FS="\t"; OFS="\t"} {print $1, $2, $6, $7, $3, $4, $5, $8}' $VIRSORTER/WATER-all_gene_calls_TEMP.txt > $VIRSORTER/WATER-all_gene_calls.txt
awk 'BEGIN {FS="\t"; OFS="\t"} {print $1, $2, $6, $7, $3, $4, $5, $8}' $VIRSORTER/MAT-all_gene_calls_TEMP.txt > $VIRSORTER/MAT-all_gene_calls.txt
#
# ----------------run virsorter command for anvio import------------------- #
#
python $VIRSORTER/helper_scripts/virsorter_to_anvio.py --db 2 -a $VIRSORTER/WATER/Metric_files/VIRSorter_affi-contigs.tab -g $VIRSORTER/WATER/VIRSorter_global-phage-signal.csv -s $VIRSORTER/WATER-splits_basic_info.txt -n $VIRSORTER/WATER-all_gene_calls.txt -f $VIRSORTER/helper_scripts/db2_hallmark_functions.txt -A $VIRSORTER/WATER-virsorter_additional_info.txt -F $VIRSORTER/WATER-virsorter_annotations.txt -C $VIRSORTER/WATER-virsorter_collection.txt
#
# ----------------import annotation data with anvio------------------- #
#
anvi-import-misc-data $VIRSORTER/WATER-virsorter_additional_info.txt -p 06_MERGED/WATER/PROFILE.db --target-data-table items
anvi-import-collection $VIRSORTER/WATER-virsorter_collection.txt -c 03_CONTIGS/WATER-contigs.db -p 06_MERGED/WATER/PROFILE.db -C VIRSORTER
anvi-import-functions -c 03_CONTIGS/WATER-contigs.db -i $VIRSORTER/WATER-virsorter_annotations.txt
#
deactivate
#
echo = `date` job $JOB_NAME done
Conclusion
This section of the workflow is complete. Lets take a look at the what we have so far.
| Directory | Description |
|---|---|
00_RAW/ |
Raw fastq files before trimming. |
00_LOGS/ |
Individual log files for each step of the snakemake workflow. |
00_TRIMMED/ |
Eight trimmed, compressed fastq files for each sample. |
01_QC/ |
Merged forward (R1) & reverse (R2) QC’ed fastq files. QC STATS file for each sample. qc-report.txt file, a summary table of all QC results. |
02_FASTA/ |
A contig fasta file and reformat report for each assembly. |
03_CONTIGS/ |
An annotated contig database for each assembly. |
04_MAPPING/ |
Short read BAM files. |
05_ANVIO_PROFILE/ |
Individual profile database for each sample. |
06_MERGED/ |
Single merged profile database. |
Source Code
The source code for this page can be accessed on GitHub by clicking this link.
Data Availability
All files generated from this workflow and described in the final table can be downloaded from figshare at doi:10.25573/data.12808601. The contigs db contains all taxonomic and functional annotations. The profile db contains information on coverage, detection, etc. as well as COLLECTION information for automatic binning and manual refinement (see step 5).